




♦ What is Creutzfeld-Jakob (CJD)?
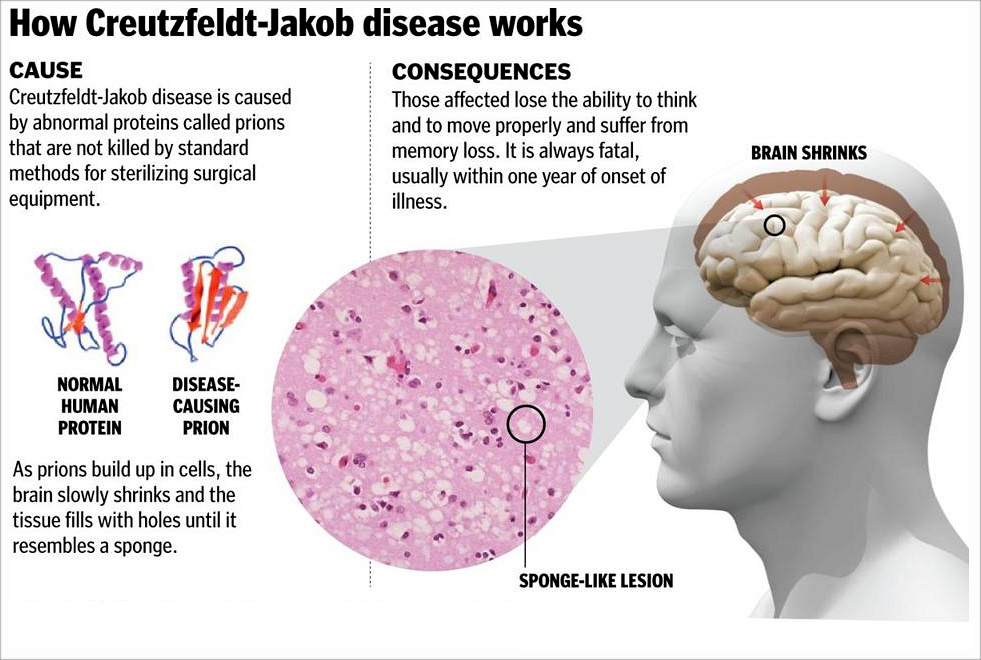
also known as classic Creutzfeldt–Jakob disease, is a fatal degenerative brain disorder.
Early symptoms include memory problems, behavioral changes, poor coordination, and visual disturbances. Later dementia, involuntary movements, blindness, weakness, and coma occur. About 90% of people die within a year of diagnosis.
CJD is believed to be caused by a protein known as a prion. Infectious prions are misfolded proteins that can cause normally folded proteins to become misfolded. Most cases occur spontaneously, while about 7.5% of cases are inherited from a person's parents in an autosomal dominant manner. Exposure to brain or spinal tissue from an infected person may also result in spread.
There is no evidence that it can spread between people via normal contact or blood transfusions.
Diagnosis involves ruling out other potential causes. An electroencephalogram, spinal tap, or magnetic resonance imaging may support the diagnosis.
There is no specific treatment. Opioids may be used to help with pain, while clonazepam or sodium valproate may help with involuntary movements.
CJD affects about one per million people per year. Onset is typically around 60 years of age. The condition was first described in 1920. It is classified as a type of transmissible spongiform encephalopathy.
CJD is different from bovine spongiform encephalopathy (mad cow disease) and variant Creutzfeldt–Jakob disease (vCJD).
♦ Sign and symptoms
• The first symptom of CJD is usually rapidly progressive dementia, leading to memory loss, personality changes, and hallucinations.
• Myoclonus (jerky movements) typically occurs in 90% of cases, but may be absent at initial onset.
• Other frequently occurring features include anxiety, depression, paranoia, obsessive-compulsive symptoms, and psychosis.
• This is accompanied by physical problems such as speech impairment, balance and coordination dysfunction (ataxia), changes in gait, rigid posture.
• In most people with CJD, these symptoms are accompanied by involuntary movements and the appearance of an atypical, diagnostic electroencephalogram tracing.
• The duration of the disease varies greatly, but sporadic (non-inherited) CJD can be fatal within months or even weeks. Most victims die six months after initial symptoms appear, often of pneumonia due to impaired coughing reflexes.
• About 15% of people with CJD survive for two or more years.
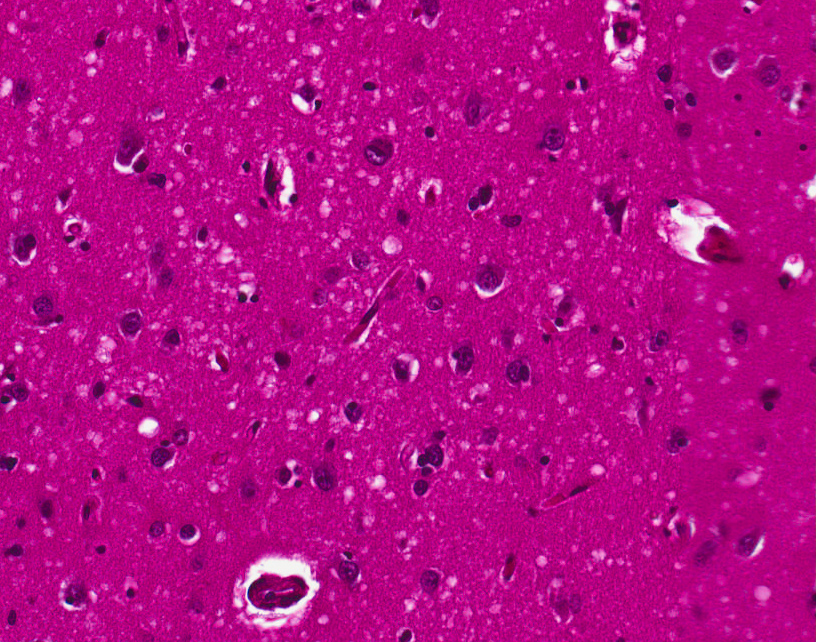
The symptoms of CJD are caused by the progressive death of the brain's nerve cells, which is associated with the build-up of abnormal prion protein molecules forming amyloids. When brain tissue from a person with CJD is examined under a microscope, many tiny holes can be seen where whole areas of nerve cells have died. The word "spongiform" in "transmissible spongiform encephalopathies" refers to the sponge-like appearance of the brain tissue.
♦ Cause
Transmissible spongiform encephalopathy diseases are caused by prions.
Prions are proteins that occur normally in neurons of the central nervous system (CNS). These proteins, once misfolded, are thought to affect signaling processes, damaging neurons and resulting in degeneration that causes the spongiform appearance in the affected brain.
The CJD prion is dangerous because it promotes refolding of native prion protein into the diseased state. The number of misfolded protein molecules will increase exponentially and the process leads to a large quantity of insoluble protein in affected cells. This mass of misfolded proteins disrupts neuronal cell function and causes cell death. Mutations in the gene for the prion protein can cause a misfolding of the dominantly alpha helical regions into beta pleated sheets. This change in conformation disables the ability of the protein to undergo digestion. Once the prion is transmitted, the defective proteins invade the brain and induce other prion protein molecules to misfold in a self-sustaining feedback loop. These neurodegenerative diseases are commonly called prion diseases.
People can also develop CJD because they carry a mutation of the gene that codes for the prion protein (PRNP). This occurs in only 5–10% of all CJD cases. In sporadic cases the misfolding of the prion protein probably occurs as a natural, spontaneous process.[13] An EU study determined that "87% of cases were sporadic, 8% genetic, 5% iatrogenic and less than 1% variant."
♦ Transmission
The defective protein can be transmitted by contaminated harvested human brain products, corneal grafts, dural grafts, or electrode implants and human growth hormone.
► It can be familial (fCJD);
► or it may appear without risk factors (sporadic form: sCJD).
In the familial form, a mutation has occurred in the gene for PrP, PRNP, in that family. All types of CJD are transmissible irrespective of how they occur in the patient.
It is thought that humans can contract the disease by consuming material from animals infected with the bovine form of the disease.
Cannibalism has also been implicated as a transmission mechanism for abnormal prions, causing the disease known as kuru, once found primarily among women and children of the Fore people in Papua New Guinea. While the men of the tribe ate the body of the deceased and rarely contracted the disease, the women and children, who ate the less desirable body parts, including the brain, were eight times more likely than men to contract kuru from infected tissue.
Prions, the infectious agent of CJD, may not be inactivated by means of routine surgical instrument sterilization procedures.
The World Health Organization and the US Centers for Disease Control and Prevention recommend that instrumentation used in such cases be immediately destroyed after use; short of destruction, it is recommended that heat and chemical decontamination be used in combination to process instruments that come in contact with high-infectivity tissues.
♦ Diagnosis
Testing for CJD has historically been problematic, due to nonspecific nature of early symptoms and difficulty in safely obtaining brain tissue for confirmation. The diagnosis may initially be suspected in a person with rapidly progressing dementia, particularly when they are also found with the characteristic medical signs and symptoms such as involuntary muscle jerking, difficulty with coordination/balance and walking, and visual disturbances. Further testing can support the diagnosis and may include:
♦ Electroencephalography – may have characteristic generalized periodic sharp wave pattern. Periodic sharp wave complexes develop in half of the patients with sporadic CJD, particularly in the later stages.
♦ Cerebrospinal fluid analysis for elevated levels of 14-3-3 protein could be supportive in the diagnosis of sCJD. However, a positive result should not be regarded as sufficient for the diagnosis. The Real-Time Quaking-Induced Conversion (RT-QuIC) assay has a diagnostic sensitivity of more than 80% and a specificity approaching 100%, tested in detecting PrPScin CSF samples of CJD patients. It is therefore suggested as a high-value diagnostic method for the disease.
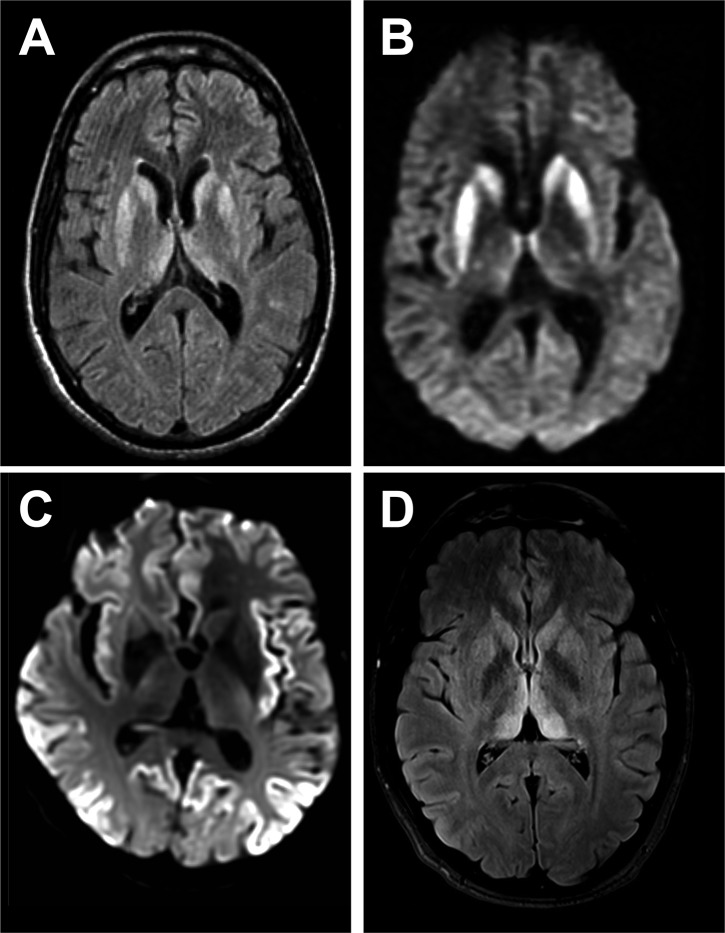
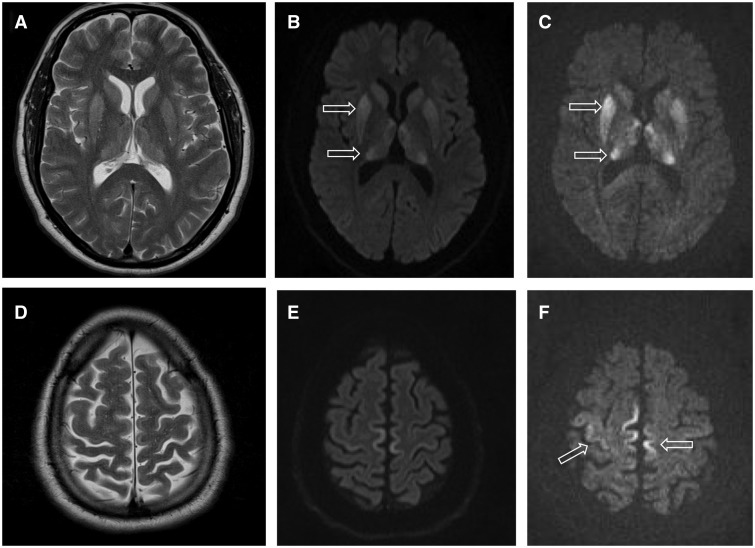
♦ MRI of the brain – often shows high signal intensity in the caudate nucleus and putamen bilaterally on T2-weighted images.
In recent years, studies have shown that the tumour marker Neuron-specific enolase (NSE) is often elevated in CJD cases; however, its diagnostic utility is seen primarily when combined with a test for the 14-3-3 protein. As of 2010, screening tests to identify infected asymptomatic individuals, such as blood donors, are not yet available, though methods have been proposed and evaluated.
♦ Histopathology
Testing of tissue remains the most definitive way of confirming the diagnosis of CJD, although it must be recognized that even biopsy is not always conclusive.
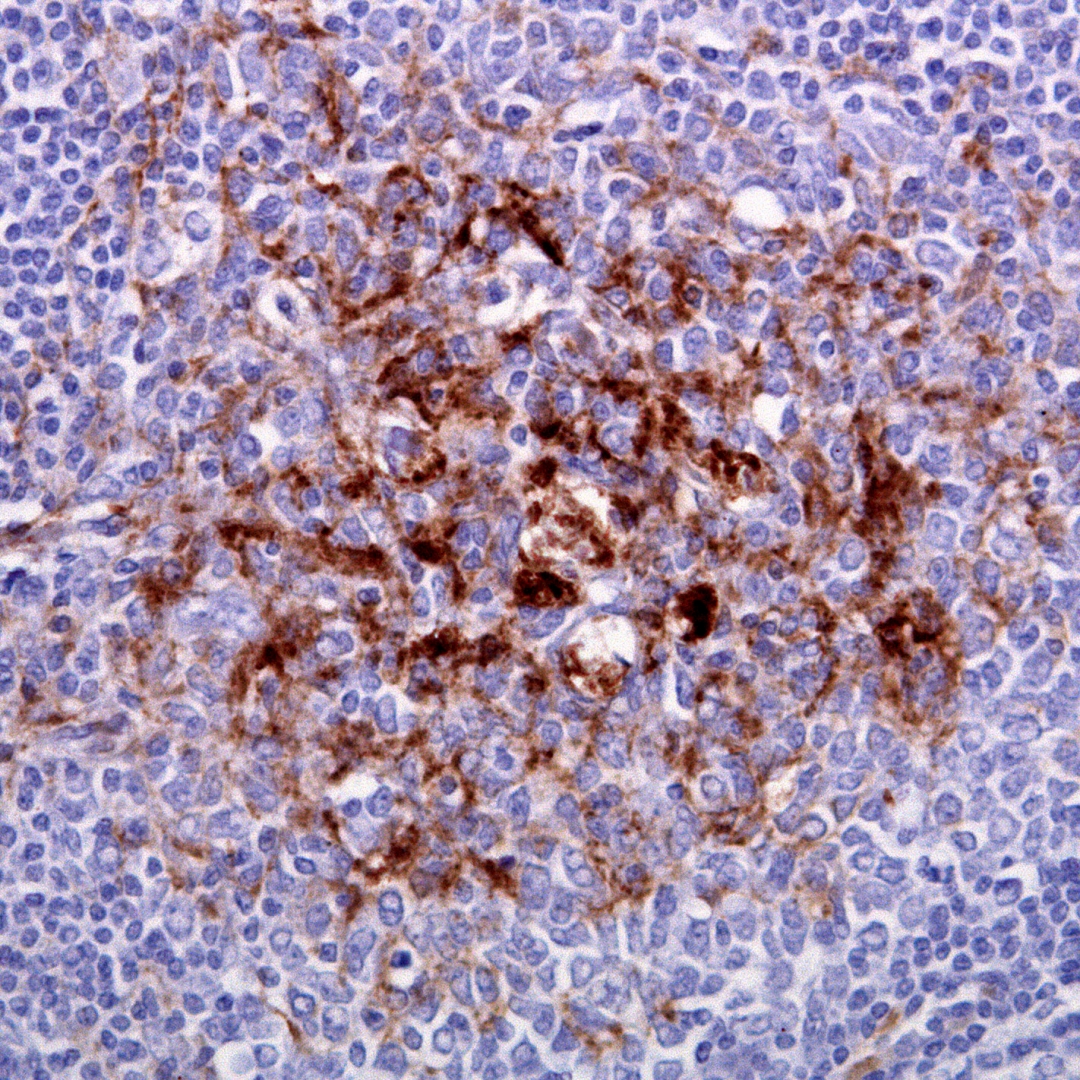
In one-third of patients with sporadic CJD, deposits of "prion protein (scrapie)," PrPSc, can be found in the skeletal muscle and/or the spleen. Diagnosis of vCJD can be supported by biopsy of the tonsils, which harbour significant amounts of PrPSc; however, biopsy of brain tissue is the definitive diagnostic test for all other forms of prion disease. Due to its invasiveness, biopsy will not be done if clinical suspicion is sufficiently high or low. A negative biopsy does not rule out CJD, since it may predominate in a specific part of the brain.
The classic histologic appearance is spongiform change in the gray matter: the presence of many round vacuoles from one to 50 micrometers in the neuropil, in all six cortical layers in the cerebral cortex or with diffuse involvement of the cerebellar molecular layer. These vacuoles appear glassy or eosinophilic and may coalesce. Neuronal loss and gliosis are also seen. Plaques of amyloid-like material can be seen in the neocortex in some cases of CJD.
However, extra-neuronal vacuolization can also be seen in other disease states. Diffuse cortical vacuolization occurs in Alzheimer's disease, and superficial cortical vacuolization occurs in ischemia and frontotemporal dementia. These vacuoles appear clear and punched-out. Larger vacuoles encircling neurons, vessels, and glia are a possible processing artifact.
♦ Classification
Types of CJD include:
♦ Sporadic (sCJD), caused by the spontaneous misfolding of prion-protein in an individual. This accounts for 85% of cases of CJD.
♦ Familial (fCJD), caused by an inherited mutation in the prion-protein gene. This accounts for the majority of the other 15% of cases of CJD.
♦ Acquired CJD, caused by contamination with tissue from an infected person, usually as the result of a medical procedure (iatrogenic CJD). Medical procedures that are associated with the spread of this form of CJD include blood transfusion from the infected person, use of human-derived pituitary growth hormones, gonadotropin hormone therapy, and corneal and meningeal transplants.
♦ Variant Creutzfeldt–Jakob disease (vCJD) is a type of acquired CJD potentially acquired from bovine spongiform encephalopathy or caused by consuming food contaminated with prions.
♦ Treatment
As of 2015 there was no cure for CJD.
Some of the symptoms like twitching can be managed but otherwise treatment is palliative care. Psychiatric symptoms like anxiety and depression can be treated with sedatives and antidepressants. Myoclonic jerks can be handled with clonazepam or sodium valproate. Opiates can help in pain. Seizures are very uncommon, and can be treated with antiepileptic drugs.

Creutzfeldt-Jakob Disease
Related
References
Wikipedia
https://www.medicalnewstoday.com/articles/185884.php
Photo
Wikipedia
- Actinomycosis
- Anthrax
- Biopsy Sinusitis_Aspergillus flavus
- Botulism
- Brucellosis
- Cat Scratch Disease
- Cellulitis
- Cholera
- Creutzfeldt-Jakob Disease
- Cystic Fibrosis_CF
- Diphtheria
- Erysipelas
- Erysipeloid or fish poison
- Legionnaires disease
- Lemierre syndrome
- Leprosy
- Listeriosis
- Lyme / Borreliosis
- Melioidosis
- Meningitis
- Plague
- Syphilis
- Tetanus
- Trench Mouth_Plaut-Vincent_acute necrotizing ulcerative gingivitis
- Tuberculosis (TB)
- Tularemia_Rabbit Fever
- Typhoid fever (Epidemic typhus)
- Whooping Cough